
Research
Carbonara
Much of my work has been based on the development of a software for ab-initio structure prediction from BioSAXS data. Biological small angle X-ray scattering is a technique for determining protein structures in solution which is useful for proteins which fail to crystallise or are too large for NMR. The original model used a novel constrained backbone algorithm to randomly search the space of possible conformations in order to fit experimental scattering data. This algorithm uses local geometric constraints on the backbone which are analogous to the Ramachandran distributions of dihedral angles. Though these constraints ensure the predicted structures are locally realistic, the global fold may not be. By studying the distribution of entanglement across the PDB, we are able to include a penalty for unrealistic global entanglement. This massively reduces the search space of possible conformations, and ensures the output structures are realistic and behave well in MD simulations. The full carbonara software package will be released for public soon, but if you have scattering data that you would like to study in the meantime please do contact us!
Writhe
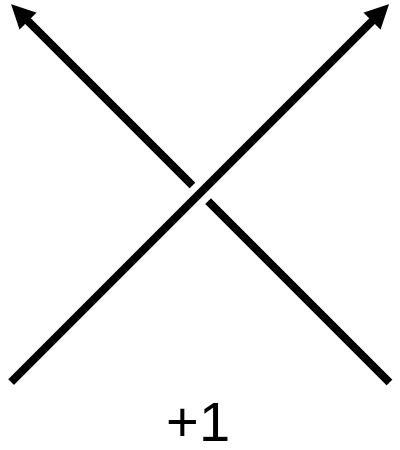
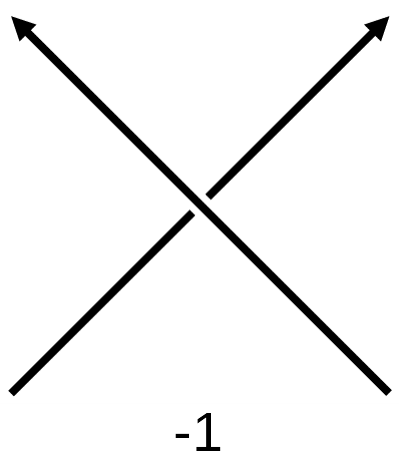
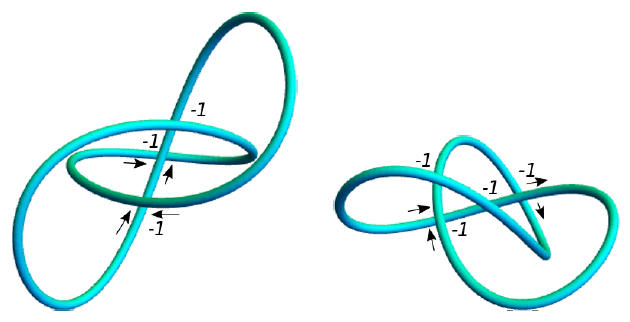
To study the entanglement of protein backbone curves we use the writhe. This is a topological quantity which measures the amount of self entanglement of a curve. This quantity has a wide range of uses, from solar physics to DNA biology. To compute this, imagine your curve is projected onto a plane. There will be points where it passes over itself, which we call crossings. We give each crossing a sign using the below convention. The signed sum of crossings will be dependent on the angle of projection, so the writhe is an average of the signed sum of crossings over all possible projections.
Publications
with Chris Prior and Rob Rambo.
We present fast and simple-to-implement measures of the entanglement of protein tertiary structures which are appropriate for highly flexible structure comparison. These are performed using the SKMT algorithm, a novel method of smoothing the alpha-carbon backbone to achieve a minimal complexity curve representation of the manner in which the protein's secondary structure elements fold to form its tertiary structure. Its subsequent complexity is characterised using measures based on the writhe and crossing number quantities heavily utilised in DNA topology studies, and which have shown promising results when applied to proteins recently. The SKMT smoothing is used to derive empirical bounds on a protein's entanglement relative to its number of secondary structure elements. We show that large scale helical geometries dominantly account for the maximum growth in entanglement of protein monomers, and further that this large scale helical geometry is present in a large array of proteins, consistent across a number of different protein structure types and sequences. We also show how these bounds can be used to constrain the search space of protein structure prediction from small angle x-ray scattering experiments, a method highly suited to determining the likely structure of proteins in solution where crystal structure or machine learning based predictions often fail to match experimental data. Finally we develop a structural comparison metric based on the SKMT smoothing which is used in one specific case to demonstrate significant structural similarity between Rossmann fold and TIM Barrel proteins, a link which is potentially significant as attempts to engineer the latter have in the past produced the former. We provide the SWRITHE interactive python notebook to calculate these metrics.
with Chris Prior.
Contributed to a chapter in the peer-reviewed book "Helicities in Geophysics, Astrophysics, and Beyond". This includes some of my earliest work on the applications of the various forms of writhing on protein structure studies.